�������ڣ�2017��3��21�� �Ķ���1821
3��17�գ�CFDA�������棬һ���ֹܾ���������ʳƷҩƷ�ල�����ֹܾ��ڵ�������ҩƷע������й�����ľ���(���������)�������֪ͨ;�����ֹܾ��ڷ�������ҩ�α��Ƽ�Ŀ¼(��*��)��ͨ��(2017���45��)��
����������2017��1�¡�����Ժ�칫�����ڽ�һ���ĸ�����ҩƷ������ͨʹ�����ߵ�������������ᵽ��Ҫ�Ż�ҩƷ�����������ӿ��ƽ������з���ҩ��������Чһ�������۵�����������һ�¡�
��Ҳ��ζ�ţ�2015��8����CFDA��ʼ��һϵ��ҩƷע��ĸ����ڼ�������Ӧ��Ŀ�����ڼ�ǿҩƷע��������ӿ�����ٴ���ֵ����ҩ���ٴ��������ҩ���з����У����ҩƷע�������ѹ��ì�ܡ����ң��ĸﲻ�����ǹ��ڻ�ѧҩ��ע��ĸ����**����Χ���쵽�˽���ҩ��ע��������
����ҩ�α��Լ�Ŀ¼����������
�Ƽ��α��Ƽ�Ʒ�ֶ������֣���ЩƷ�����ӹ�������Ϊ**�IJ�Ʒע�����Դ
3��17�գ�CFDA��������ҩ�α��Ƽ�Ŀ¼(��*��)34��Ʒ��51��Ʒ��;������3��20�ռ������˷���ҩ�α��Ƽ�Ŀ¼(�ڶ���)21��Ʒ��33��Ʒ�棬�ϼ�43��Ʒ��84��Ʒ�档
��*��CFDA������34��Ʒ�ֶ���2018�����ǰ��Ҫ���һ�������۵�289Ŀ¼�ڵIJ�Ʒ���ڶ���������Ʒ�ֳ��˶�������Ʒ������ƥ��¡Ƭ��12��Ʒ������Ҫ���һ�������۵�289Ŀ¼�ڵIJ�Ʒ�������289Ŀ¼�еIJ�Ʒͬͨ������ͬ���͵IJ�Ʒ����ͷ�������ɻ����������ᰱ�������ͽ��ҡ�
��CDE������2016���ҩƷ�������桷��Ŀǰ����ҩһ��������BE����Ʒ��16�����̴�����V3.2���֣� BE�����������ٴ������ҵǼǺ�Ϊ2016��IJ�Ʒ����������������Ƭ�����ᰢ������Ƭ�����ᰬ˾��̪����Ƭ�������ᰱ�ȵ�ƽƬ�������ᰢ����͡Ƭ����������������Ƭ���ױ�������������Ƭ����������������Ƭ����ɳ������Ƭ����������Ƭ���������Ƭ������һ��ͽ��ҡ���������ŵ��Τ������Ƭ��������Ƭ������ɳ��Ƭ����������Ƭ������Щ��Ʒ���Ƕ���һ��������BE����Ʒ�֣����б����ᰱ�ȵ�ƽƬ����������������Ƭ����������ƬΪ289Ŀ¼�ڲ�Ʒ����������������Ƭ��������Ƭ��CFDA����ҩ�α��Ƽ�Ŀ¼�С�
����2017��3��20�գ��й�ʳƷҩƷ�춨�о�Ժ�ֱ���2016��8�¡�9�º�11�·ֱ��Ƽ��α��Ƽ�Ʒ�֣��ϼ��Ƽ�17��Ʒ��(12��Ʒ��)������Ʒ��������3��17�յġ�����ҩ�α��Ƽ�Ŀ¼(��*��)��51��Ʒ�����������֣�����ͷ������Ƭ���м�Ժ���Ƽ�������ֻ��0.25g���CFDA��*��Ŀ¼�Ƽ�����0.125g��ڶ���Ŀ¼����������0.5g���
������ҩ�α��Ƽ�Ŀ¼(�ڶ���)�����й�ʳƷҩƷ�춨�о�Ժ���Ƽ�ԭ�е�Ʒ����ͬͨ���������ӹ�����������͡Ƭ������5mg��40mg�����������ƽƬ������0.2g��0.3g��50mg�������������Ƭ������1mg��5mg����������������Ƭ������25mg����������������Ƭ����300mg��
ֵ��ע����ǣ�CFDA�����ķ���ҩ�α��Ƽ�Ŀ¼��������Ʒ��Ϊ**��ע�����Դ����֪���Ƿ���ζ�Ź��ڷ���ҩ����������Ҫ������Դ���ڹ�ѡ����α��Ƽ�?������ʵ������ζ�ű�1��34��Ʒ��53Ʒ�����ѡ����ڽ��ڲ�Ʒ���α��Ƽ���
��1 ��ͬ��Դ��Ӧ�IJ�Ʒ��

������Դ���̴�����V3.2
�̴�����V3.2���֣� 1�¡�����Ժ�칫�����ڽ�һ���ĸ�����ҩƷ������ͨʹ�����ߵ�����������������м�Ժ��2��9�շ���������FDA��Ƥ��(�������Ƶ�Ч����������ҩƷ)���ĺ��ձ���Ƥ��(ҽ����ҽҩƷƷ���鱨��)���ġ���������CFDA���м�ԺΪ�ƶ�һ�������۶�������Ϣ�������ƽ���Ŭ����
����ע���������Լӿ�
���ڴ�ǰ���ý���ҩ�걨�Ŷ�ʱ��ϳ������µľ�3.1����ҵ����ϴ�
CFDA����Ҫ�����ҩ��������ҵ�й��������ҩƷ��Ʒ�Ľ������̾�Ҫ��ͬʱ�ύ��������֤�������ע�����룬ȷ�ϸ�ҩƷ���ڸù����в�����֮���������й����ٴ�������Ȩ���⽫����**��ҩ�����й����е�ʱ�䣬Ҳʹ���й��Ļ�����ʹ��ȫ���г�*�ߴ����Ե�ҩ����֮�£����������ң�ԭ�����ٴ��������������ٴ�����ͬʱ���С�
��2 �Ա�ԭҩƷע������취
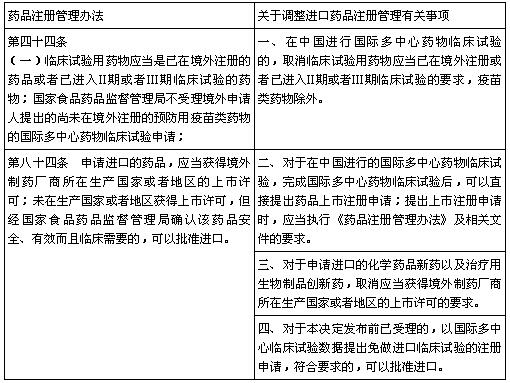
(������Դ���̴�����V3.2)
���վɻ�ѧҩע����࣬��������ҩ��ע������1�����3�࣬��Ӧ�ֻ�ѧҩ�·����5.1���5.2�ࡣ���о������������й����й��ʶ�����ҩ���ٴ�����IMCT(International Multicenter Clinical Trial)�Ļ���Ҫ�����ʳƷҩƷ�ල������������롣
����IMCTҩ����ԣ���Ϊ��ҩ��û�о�����ҩ���������������һ��ߵ�������������֤��CPP(certificate of a pharmaceutical product)���������3��ҩ�����ǣ������걨����1��ҩ�������������˵����������������Ҫ�Ƚ���һ���ٴ����ɵ����룬Ȼ�������һ�������Ģ��ڡ����ڡ������ٴ����飬��������������������ҩ����(NDA)��*��õ�����ҩƷע��֤(IDL)��
�����������Ģ���������ʱ��ͷ��óɱ����ϸߣ�����*��Ҫ��CPP��������걨��������ˣ������걨����ּ�ڹ����ѻ��CPP��ǰ���£��ڹ����걨IMCT�IJ����ٴ����������a����b�����ڵ�PK������ʹ�á�������ҩ���Ա��о������ܶ���֮��ͬ����Ʒ����������ͬĿ�ĵ�IMCT��ö���ٴ�CTP�ǼǺš�������Ŀ�����Ծ��dz��������й��Խ���ҩƷ3���ٴ�����Ҫ��ȥ�ġ�����ɸ�������Խ���ҩƷ3����ٴ��������ȡ����CDE�Ĵ���ҩ�����������϶�IMCT�������������������̱�NDA���ٴ���ֱ�ӻ��IDL��
��ν�������������������ա�IMCT����-�ٴ�����-JXHL����(���ٴ�)-ͬ��NDA���ٴ������IDL�������ij��������ֳ����������������IMCT���ٴ�CTP�ǼǺ�Ԥ����Ҫ10��������(����������������)���ٴ�����3~5�꣬��֤���ٴ���������(���ٴ�)��Ҫ24~26���£��ֹ۹��ƣ����걨IMCT�����IDL����Ҫ8~10�ꡣ
����IMCTԽ��Խ�࣬��ҩ�ٴ���������(IND)�Ļ�ѹ��ʼԽ��Խ�࣬CDE������ʱ��ҲԽ��Խ����CFDA/CDE����Ҫ�����ҩƷע������ϸ��ա�ҩƷע������취���涨�ij�������������������IMCT����-�ٴ�����-JXHL������ҩ��ѧҩ�ٴ���������(���ٴ�)-������ҩ��ѧҩ����JXHS����-��ý���ҩƷע��֤IDL�������ij�����CFDA/CDE�������ٶȣ���������������ζ��������2.5���������õ�����ʱ�䣬ʹ��IMCT;��ȡ�õ�IDL�ƺ�2���������á�
CFDA���θĸ�������Ҫ�Ǹı����»�ѧҩע�����5.1���ע�����IMCT�Ģ����ٴ����п���Ҳ�������й����ٴ�������������Ҫ���ǣ�Ŀǰ���Ŷ��걨������������ѹ�������ٴ��Ľ���ҩ�����ڽ��ڿ������У�ǰ�����ٴ��Բ�˲�Ҫ���ء�
���¹���ڹ�����ҵ��ǰ���ڽ���ҩ���걨�Ŷ�ʱ������Ӷ����¾�3.1��ҩƷ����ҵ����ϴ����ȣ�ԭ��ҩ�����ڽ���ȫ��������ζ������ԭ��ҩǰ���л���������;�����ԭ��ҩ��ר���ڱ����£�2015����2016�����������ٴ��Ĺ��ڷ���ҩ��ҵ��ʹ��2016����2017�������ٴ�������ҲҪ��ר����ǰ1���걨���������ң������¹棬����ҩ��Ҫ�����һ�����������飬ԭ���걨��3.1�����ҵ������ֱ����BE���������ŶӸ����ʡǮ��
�ܽ�>>>
������һ����������Ϣ���������ǽ���ҩƷע������й�����ĵ�����������CFDA��ҩƷע��������ѹ�ĸ�Ļ���̬�ȡ�3��15��ҩ�����ķ����ӿ��ƽ�eCTD��Ŀ����Ĺ��棬��ζ��2017��������л�ѧ����ҩ���걨����������eCTDϵͳ��ʵʩ�Ƿdz��п��ܵġ�CFDA��CDE��Ŀ���������ʻ�������������һ���ĸĸ����ȫ��ѧϰFDA��ȡ����ҩ�P���Ƽ�������(NDA)������ҩ������(ANDA)��OTC(�Ǵ���ҩ)������ģʽ��
���⣬��ѧҩ�ĸ���ɺ���ҩ��������Ʒ�ĸĸ�Ҳ����֮������Ŀǰֻ�ǽ��Ըĸ������ҵ��������ʱ�̽�������ҩƷע�����ߵ�����֮Ϊ��Ҫ���ɣ���Ӧ�ų�Ŀ�⣬���������ʻ��ͺϹ滯����չ��Ϊ������
������Դ��
1.������ע������Դ��1168ҽҩ����������������Ʒ����Ϊ���ݽ��ڻ������Ƽ�����˾-1168ҽҩ�������Ϸ�ӵ�а�Ȩ����Ȩʹ�õ���Ʒ��δ��������Ȩ����ת�ء�ժ�������������ʽʹ��������Ʒ���Ѿ�������Ȩʹ����Ʒ�ģ�Ӧ����Ȩ��Χ��ʹ�ã���ע������Դ��1168ҽҩ������http://www.1168.tv����Υ�����������ߣ�������������ط������Ρ�
2.����ת�ز�ע����������Դ����1168ҽҩ������������Ʒ��Ŀ�����ڴ��ݸ�����Ϣ����������������ͬ��۵��Ͷ�����ʵ�Ը��𣬲��е�������Ʒ��Ȩ��Ϊ��ֱ�����μ��������Ρ�
3.����ý�塢��վ����˴ӱ���ת��ʱ�����뱣������ע������Ʒ��һ��Դ�����Ը���Ȩ�ȷ������Ρ�
4.���漰��Ʒ���ݡ���Ȩ�����⣬������Ʒ����֮����һ�����뱾����ϵ��������Ϊ�������Ȩ������ϵ���䣺1753418380@qq.com��

�����÷�Χ�����ڻ��⾱���硢�����ȼ��պ�������֯��ʹ�����͵Ȳ���֢״��Ⱥ���������ʹ�÷��������á�����Ʒ����ֱ��ͿĨ�ڲ��ʲ�λ�����ᰴĦ2-3���ӣ�ÿ��2-3�Ρ�

�����÷�Χ�����ڻ��⾱���硢�����ȼ��պ�������֯��ʹ�����͵Ȳ���֢״��Ⱥ���������ʹ�÷��������á�����Ʒ����ֱ��ͿĨ�ڲ��ʲ�λ�����ᰴĦ2-3���ӣ�ÿ��2-3�Ρ�








 ���������� 44011102000390��
���������� 44011102000390��